Layla Oesper
Research
Cancer is a disease resulting from genomic mutations that occur during an individual's lifetime and cause the uncontrolled growth of a collection of cells into a tumor. As we enter the era of personalized medicine, where a patient's treatment may be tailored to their specific genomic architecture, accurate identification and interpretation of the set of mutations within each patient's genome is increasingly important. Inspired by this need, my research focuses on designing algorithms that enable analysis and interpretation of high-throughput DNA sequencing data of cancer genomes.
Intra-tumor Heterogeneity and Tumor Evolution
Tumors often exhibit intra-tumor heterogeneity where individual cells in a single tumor contain different complements of mutations. This heterogeneity arises because tumors evolve overtime as descendant cells acquire new somatic mutations. This is further complicated as tumors typically also contain admixture with normal (healthy) cells. We do not observe this evolutionary history directly, but instead must infer it from data obtained at the time a patient is diagnosed.
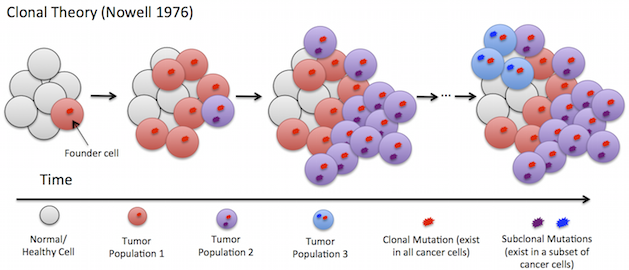
I have worked on methods to infer the evolutionary history of heterogeneous tumors as a type of rooted tree called a clonal tree. AncesTree and SPRUCE are algorithms that infer the clonal evolution and tumor composition of a tumor from multi-sample DNA sequence data.
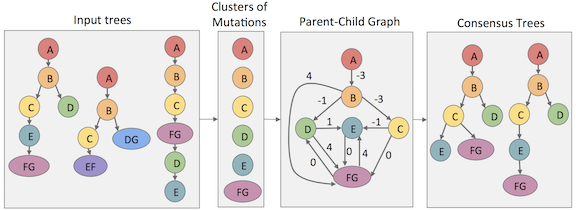
More recently my work has focused on ways to make the clonal trees inferred by such methods more accurate and useful. GraPhyC is a graph-based algorithm that takes in a set of potential tumor evolutionary histories and finds a consensus (or representative) history. TuELiP is a recent consensus method that allows all input trees to be weighted differently. CASet and DISC are distance measures to compare two inferred clonal trees that was recently developed in my lab. Such distance measures will be an essential part to improving benchmarking procedures for new clonal tree inference methods.
Previously, I designed methods to infer the composition of heterogeneous tumor samples from bulk DNA sequencing data. For instance, THetA/THetA2 is an algorithm that estimates tumor purity and clonal/subclonal copy number aberrations directly from high-throughput DNA sequencing data.
Complex Rearrangements
Cancer genomes are highly rearranged and often contain complex rearrangement patterns that amplify, delete or interweave distant regions of the genome. These complex rearrangement patterns make it difficult to identify the true set of rearrangements that exist in a genome.
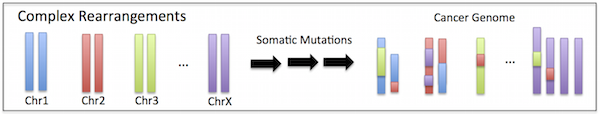
I have designed several methods to better enable analysis of cancer genomes that contain complex rearrangements. PREGO is an efficient network flow inspired algorithm that finds the collection of rearrangements that together best describe the observed sequence data, rather than treating each rearrangement as an independent event. Furthermore, I was also involved in the development of two measures: (1) the open adjacency rate (OAR); (2) copy-number asymmetry enrichment (CAE), that assess the prevalence of simultaneously formed rearrangements from sequencing data of a cancer genome as the result of events such as chromothripsis. More recently, I developed the H/T Alternating Fraction measure of chromothripsis.